Case Report
Clinical Findings
Een 65-jarige vrouw presenteerde zich met meerdere asymptomatische discrete noduli en atrofische plaques op de dijen die al 4 jaar bestonden. De laesies waren begonnen als 2 kleine, asymptomatische, roodbruine plaques symmetrisch gelegen op het voorste deel van elk dijbeen die geleidelijk in aantal en grootte waren toegenomen, vooral op het rechter dijbeen. Twee jaar later verschenen 2 nieuwe atrofische plaques aan de voorkant van elk van beide dijen. De letsels ontwikkelden zich langzaam maar werden nooit minder en waren door verschillende poliklinieken verkeerd gediagnosticeerd als primaire maculaire atrofie van de huid. De algemene gezondheidstoestand van de patiënte was goed en haar persoonlijke en familiale voorgeschiedenis was onopvallend.
Bij lichamelijk onderzoek werden op het voorste deel van de rechterdij meerdere, rossige plaques en knobbels van verschillende vorm en grootte (d.w.z. 1-3 cm in diameter) gevonden. De laesies waren licht verheven met een wasachtig oppervlak, stevig, en pijnloos bij palpatie. Eén gelijkaardige laesie werd genoteerd op het voorste deel van de linkerdij. Twee 2-cm, bruin-rode, atrofische plaques werden ook genoteerd in een symmetrische verdeling op het voorste aspect van elk dijbeen. De oppervlakken van de plaques waren enigszins rimpelig en glanzend, en anetodermale laesies produceerden een knoopsgatenpatroon identiek aan een neurofibroom bij palpatie (Figuur 1).
 Figuur 1. Meerdere meekrap rode plaques, knobbeltjes, en atrofische plaques in verschillende vormen en maten werden gezien op het voorste aspect van beide dijen.
Figuur 1. Meerdere meekrap rode plaques, knobbeltjes, en atrofische plaques in verschillende vormen en maten werden gezien op het voorste aspect van beide dijen. Histopathologische bevindingen
Twee biopsiemonsters werden genomen van een nodule en een atrofische plaque op de rechter dij. Microscopisch onderzoek onthulde afzetting van homogeen eosinofiel materiaal in de reticulaire dermis en subcutis, evenals rond de fijne bloedvaten (Figuur 2A). Er was een milde cellulaire infiltratie van lymfocyten, plasmacellen en reuscellen in de lederhuid, vooral naast de afzettingen en rond de bloedvaten (Figuur 2B). Het homogene materiaal verscheen zalmroze door Congo rood kleuring en helder groen door thioflavine T kleuring met een fluorescente microscoop (Figuren 3 en 4). Deze resultaten suggereerden de karakteristieke kenmerken van cutane nodulaire amyloïdose.
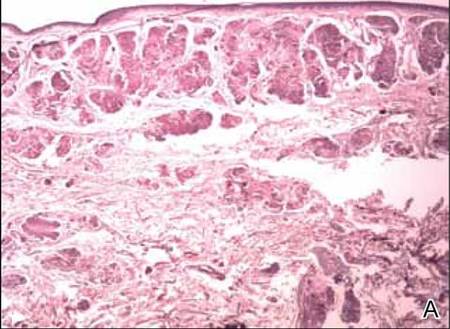
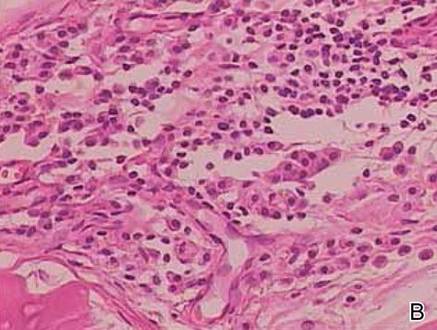
Figuur 2. Histopathologisch werd homogeen eosinofiel materiaal afgezet in de reticulaire dermis en subcutis waargenomen (A)(H&E, oorspronkelijke vergroting ×25). Microscopisch onderzoek toonde lymfocyten en plasmacellen geïnfiltreerd in de dermis, vooral grenzend aan de afzettingen en rond de vaten (B)(H&E, originele vergroting ×200).
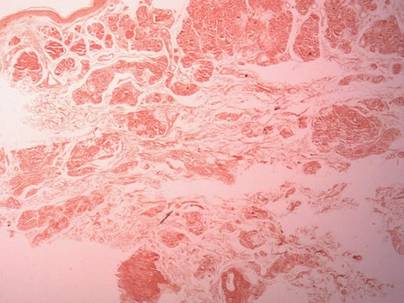
Figuur 3. De Congo rood kleuring toonde zalmroze homogeen materiaal (originele vergroting ×25).
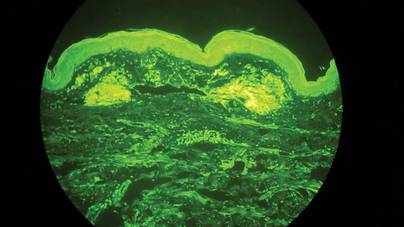
Figuur 4. Thioflavine T-kleuring toonde heldergroen homogeen materiaal (originele vergroting ×100).
Laboratoriumbevindingen
Laboratoriumonderzoek toonde normale resultaten voor volledig bloedceltelling, urineonderzoek, lever- en nierfunctietests, bloedglucosespiegels, lipidenpanel en erytrocytensedimentatiesnelheid. Serum proteïne elektroforese was normaal en er werden geen Bence Jones proteïnen gedetecteerd. Serum IgA, IgG, en IgM niveaus toonden geen afwijkingen. Elektrocardiogram, radiografie van de borst en echografie van de buik waren normaal.
De diagnose van primaire gelokaliseerde cutane nodulaire amyloïdose (PLCNA) werd gesteld op basis van klinische, histopathologische en laboratoriumbevindingen. Hoewel chirurgische excisie in fasen werd voorgesteld, weigerde de patiënte behandeling omdat de laesies asymptomatisch waren. Er was geen duidelijke progressie van de huidlaesies en geen abnormale systemische bevindingen gedurende 2,5 jaar follow-up.
Commentaar
Amyloïdose is een spectrum van ziekten bestaande uit afzetting van amyloïde eiwitten in verschillende weefsels. Klinisch wordt amyloïdose onderverdeeld in zowel primaire als secundaire vormen van systemische amyloïdose, hemodialyse-geassocieerde amyloïdose, heredofamiliaire amyloïdose, en cutane amyloïdose. Primaire cutane amyloïdose is gelokaliseerd in de huid zonder betrokkenheid van andere organen en komt niet voor bij systemische amyloïdose. Secundaire cutane betrokkenheid bij systemische amyloïdose is zeldzaam. De meeste gevallen van primaire gelokaliseerde cutane amyloïdose (PLCA) zijn sporadisch, maar ongeveer 10% van de gevallen kan familiaal zijn.1 Er zijn 3 hoofdvormen van gelokaliseerde cutane amyloïdose: maculaire, lichen, en nodulaire amyloïdose. Nodulaire cutane amyloïdose is de zeldzaamste vorm van PLCA.
Nodulaire amyloïdose werd voor het eerst beschreven door Gottron in 1950.2 De cutane laesies kunnen zich presenteren als enkelvoudige of meervoudige knobbeltjes, soms met daarboven atrofische plaques. De letsels bestaan uit stevige, gladde, wasachtige of rubberachtige, roze tot geelbruine papels, plaques of knobbels die tot enkele centimeters groot kunnen zijn. Op sommige laesies kan oppervlaktelangiëctasie worden gezien. Er zijn bulleuze en anetodermale laesies gerapporteerd.3 De acrale regio is de meest voorkomende locatie, gevolgd door respectievelijk de benen, het hoofd, de romp, de armen en de genitaliën.4 In sommige gevallen kunnen de laesies na verloop van tijd spontaan verbeteren. Bij onze patiënte bestonden de laesies uit zowel multipele noduli als atrofische plaques, wat ongewoon is.
De pathogenese van amyloïdafzetting is nog onbekend. Cutane maculaire en lichen amyloïdose kan afkomstig zijn van gedegenereerde keratinocyte intermediaire filamenten. Nodulaire amyloïdose kan een gelokaliseerde plasmacel dyscrasie vertegenwoordigen die geassocieerd kan zijn met een monoklonale gammopathie of multipel myeloom.5 Sommige componenten van amyloïd in sommige gevallen van PLCNA kunnen bestaan uit κ en λ immunoglobuline lichte ketens, waarbij de meeste gerapporteerde gevallen van het l-subtype zijn.6 De resultaten van één studie wezen uit dat β2-microglobuline een andere belangrijke component van amyloïde fibrillen was en dat β2-microglobuline gedeeltelijk onderhevig was aan de modificatie van advanced glycation end product in PLCNA.7
Geef een antwoord