Fallbericht
Klinischer Befund
Eine 65-jährige Frau stellte sich mit mehreren asymptomatischen diskreten Knötchen und atrophischen Plaques an den Oberschenkeln vor, die seit 4 Jahren bestanden. Die Läsionen begannen als zwei kleine, asymptomatische, krapprote Plaques, die sich symmetrisch auf der Vorderseite jedes Oberschenkels befanden und allmählich an Zahl und Größe zugenommen hatten, insbesondere am rechten Oberschenkel. Zwei Jahre später traten an der Vorderseite beider Oberschenkel 2 neue atrophische Plaques auf. Die Läsionen entwickelten sich langsam, bildeten sich aber nie zurück und wurden von mehreren Ambulanzen fälschlicherweise als primäre makuläre Atrophie der Haut diagnostiziert. Der allgemeine Gesundheitszustand der Patientin war gut, und ihre persönliche und familiäre Vorgeschichte war unauffällig.
Bei der körperlichen Untersuchung zeigten sich auf der Vorderseite des rechten Oberschenkels mehrere krapprote Plaques und Knötchen unterschiedlicher Form und Größe (d. h. mit einem Durchmesser von 1 bis 3 cm). Die Läsionen waren leicht erhaben, hatten eine wachsartige Oberfläche, waren fest und schmerzlos beim Abtasten. Eine ähnliche Läsion wurde an der Vorderseite des linken Oberschenkels festgestellt. Zwei 2 cm große, braun-rote, atrophische Plaques wurden ebenfalls in symmetrischer Verteilung an der Vorderseite beider Oberschenkel festgestellt. Die Oberflächen der Plaques waren leicht kraus und glänzend, und die anetodermalen Läsionen erzeugten bei der Palpation ein Knopflochzeichen, das mit einem Neurofibrom identisch war (Abbildung 1).
 Abbildung 1. Mehrere krapprote Plaques, Knötchen und atrophische Plaques in verschiedenen Formen und Größen wurden an der Vorderseite beider Oberschenkel festgestellt.
Abbildung 1. Mehrere krapprote Plaques, Knötchen und atrophische Plaques in verschiedenen Formen und Größen wurden an der Vorderseite beider Oberschenkel festgestellt. Histopathologischer Befund
Zwei Biopsieproben wurden von einem Knötchen und einer atrophischen Plaque am rechten Oberschenkel entnommen. Die mikroskopische Untersuchung ergab Ablagerungen von homogenem eosinophilem Material in der retikulären Dermis und Subkutis sowie um die feinen Gefäße (Abbildung 2A). Die Dermis wies eine leichte zelluläre Infiltration mit Lymphozyten, Plasmazellen und Riesenzellen auf, insbesondere in der Nähe der Ablagerungen und um die Gefäße (Abbildung 2B). Das homogene Material erschien bei der Kongorot-Färbung lachsfarben und bei der Thioflavin-T-Färbung unter dem Fluoreszenzmikroskop hellgrün (Abbildungen 3 und 4). Diese Ergebnisse wiesen auf die charakteristischen Merkmale einer kutanen nodulären Amyloidose hin.
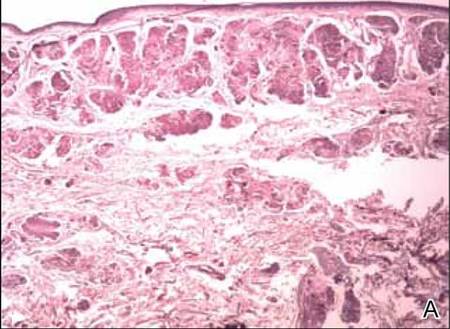
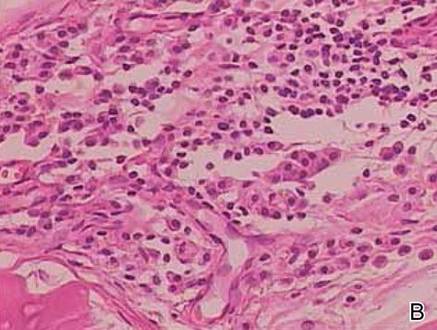
Abbildung 2. Histopathologisch wurde homogenes eosinophiles Material festgestellt, das sich in der retikulären Dermis und Subkutis ablagerte (A)(H&E, Originalvergrößerung ×25). Die mikroskopische Untersuchung zeigte Lymphozyten und Plasmazellen, die in die Dermis infiltriert waren, insbesondere in der Nähe der Ablagerungen und um die Gefäße (B)(H&E, Originalvergrößerung ×200).
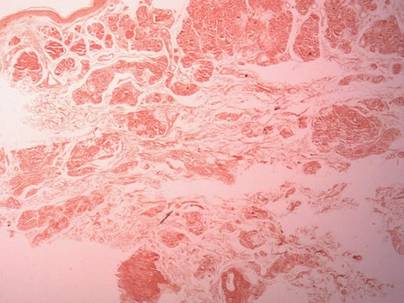
Abbildung 3. Kongorot-Färbung zeigte lachsrosa homogenes Material (Originalvergrößerung ×25).
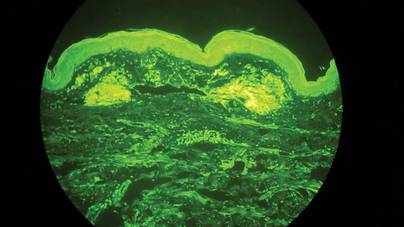
Abbildung 4. Die Thioflavin-T-Färbung zeigte hellgrünes, homogenes Material (Originalvergrößerung ×100).
Laborbefunde
Laboruntersuchungen ergaben normale Ergebnisse für das komplette Blutbild, die Urinanalyse, Leber- und Nierenfunktionstests, Blutzuckerspiegel, Lipidpanel und Erythrozytensedimentationsrate. Die Serumprotein-Elektrophorese war normal, und es wurden keine Bence-Jones-Proteine nachgewiesen. Die IgA-, IgG- und IgM-Werte im Serum zeigten keine Auffälligkeiten. Elektrokardiogramm, Röntgenaufnahme des Brustkorbs und abdominale Ultraschalluntersuchung waren normal.
Auf der Grundlage der klinischen, histopathologischen und Laborbefunde wurde die Diagnose einer primären lokalisierten kutanen nodulären Amyloidose (PLCNA) gestellt. Obwohl eine schrittweise chirurgische Exzision vorgeschlagen wurde, lehnte der Patient die Behandlung ab, da die Läsionen asymptomatisch waren. Während der 2,5-jährigen Nachbeobachtungszeit kam es zu keinem offensichtlichen Fortschreiten der Hautläsionen und zu keinen abnormen systemischen Befunden.
Kommentar
Amyloidose ist ein Spektrum von Krankheiten, die aus Ablagerungen von Amyloidproteinen in verschiedenen Geweben bestehen. Klinisch wird die Amyloidose in primäre und sekundäre Formen der systemischen Amyloidose, der Hämodialyse-assoziierten Amyloidose, der heredofamiliären Amyloidose und der kutanen Amyloidose unterteilt. Die primäre kutane Amyloidose ist auf die Haut beschränkt, ohne dass andere Organe betroffen sind, und kommt bei der systemischen Amyloidose nicht vor. Eine sekundäre kutane Beteiligung bei systemischer Amyloidose ist selten. Die meisten Fälle von primärer lokalisierter kutaner Amyloidose (PLCA) treten sporadisch auf, aber etwa 10 % der Fälle können familiär bedingt sein.1 Es gibt drei Hauptformen der lokalisierten kutanen Amyloidose: makuläre, lichenartige und noduläre Amyloidose. Die noduläre kutane Amyloidose ist die seltenste Form der PLCA.
Die noduläre Amyloidose wurde erstmals 1950 von Gottron beschrieben.2 Ihre kutanen Läsionen können als einzelne oder multiple Knötchen auftreten, gelegentlich mit darüber liegenden atrophischen Plaques. Die Läsionen bestehen aus festen, glatten, wachsartigen oder gummiartigen, rosafarbenen bis hellbraunen Papeln, Plaques oder Knötchen von bis zu mehreren Zentimetern Größe. Bei einigen Läsionen können oberflächliche Teleangiektasien zu sehen sein. Es wurde über bullenartige und anetodermale Läsionen berichtet.3 Die häufigste Lokalisation ist die Akrenregion, gefolgt von Beinen, Kopf, Rumpf, Armen und Genitalien.4 In einigen Fällen können sich die Läsionen im Laufe der Zeit spontan zurückbilden. Bei unserem Patienten bestanden die Läsionen sowohl aus multiplen Knötchen als auch aus atrophischen Plaques, was ungewöhnlich ist.
Die Pathogenese der Amyloidablagerungen ist noch unbekannt. Die kutane makuläre Amyloidose und die Lichen-Amyloidose können von degenerierten Intermediärfilamenten der Keratinozyten herrühren. Bei der nodulären Amyloidose kann es sich um eine lokalisierte Plasmazelldyskrasie handeln, die mit einer monoklonalen Gammopathie oder einem multiplen Myelom assoziiert sein kann.5 In einigen Fällen von PLCNA können einige Amyloidkomponenten aus κ- und λ-Immunglobulin-Leichtketten bestehen, wobei die meisten gemeldeten Fälle vom l-Subtyp sind.6 Die Ergebnisse einer Studie deuten darauf hin, dass β2-Mikroglobulin ein weiterer Hauptbestandteil von Amyloidfibrillen ist und dass β2-Mikroglobulin teilweise der Modifikation von fortgeschrittenen Glykierungsendprodukten in PLCNA unterworfen ist.7
Schreibe einen Kommentar