Schlüsselwörter
CADASIL – Schlaganfall – MRT
Einführung
Cerebrale autosomal dominante Arteriopathie mit subkortikalen Infarkten und Leukoenzephalopathie (CADASIL) ist eine erbliche Erkrankung der kleinen Arterien, die durch verschiedene pathogenetische Mutationen des NOTCH-3-Gens verursacht wird. Sie ist durch die Assoziation von Migräne mit Aura, psychiatrischen Symptomen, wiederkehrenden ischämischen Ereignissen in jungen Jahren und kognitiven Beeinträchtigungen gekennzeichnet. Dies und die typischen radiologischen Befunde sind es, die uns in der Regel den Verdacht auf diese Krankheit nahelegen. In der Regel wird sie als Erkrankung junger und mittelalterlicher Erwachsener angesehen, doch können Fälle bei älteren Menschen unterdiagnostiziert werden. Wir berichten über den Fall eines Mannes, bei dem in seinen späten siebziger Jahren dank der vorherigen Diagnose seiner Tochter CADASIL diagnostiziert wurde.
Fallberichte
Fall 1
Die Tochter unseres Patienten war 50 Jahre alt, als sie von der neurologischen Ambulanz zur weiteren Untersuchung in unser Krankenhaus überwiesen wurde, und zwar aufgrund der radiologischen Befunde in ihrer MRT-Aufnahme des Gehirns, die Veränderungen der weißen Substanz in beiden äußeren Kapseln, in den bilateralen periventrikulären Bereichen und im linken Temporallappen zeigten.
Sie suchte die vorgenannte Klinik auf, weil sie seit 30 Jahren an Migräne mit visueller Aura litt, wobei einige Episoden eine visuelle Aura ohne Kopfschmerzen aufwiesen. Sie hatte keine anderen Symptome. Sie war allergisch gegen Phosphomycin und hatte sich als Kind einer Tonsillektomie unterzogen. Sie hatte keine andere medizinische Vorgeschichte. Ihre Familienanamnese war bemerkenswert: Ihre Mutter litt an Migräne und leichter Demenz, ihr Vater hatte im Alter von 75 Jahren einen Schlaganfall erlitten; ihr Großvater väterlicherseits, ihr Onkel und ihre Tante hatten Schlaganfälle erlitten, und bei dem Onkel war Alzheimer diagnostiziert worden, ein Cousin väterlicherseits hatte an anderer Stelle die Diagnose „ANA+ Vaskulitis“ erhalten, und bei einem anderen Cousin väterlicherseits war Multiple Sklerose diagnostiziert worden. Sie hatte zwei gesunde Söhne. Die körperliche Untersuchung, einschließlich einer sorgfältigen neurologischen Exploration, war völlig normal. Die wiederholte MRT-Untersuchung des Gehirns zeigte multiple hyperintense Läsionen in der weißen Substanz der hinteren Protuberanz, in periventrikulären und subkortikalen Bereichen, einschließlich der vorderen Temporallappen, insbesondere des linken, beider Centrum semiovale, interner und externer Kapseln, ohne Anzeichen für aktuelle oder frühere Mikroblutungen (Abbildung 1). Alle anderen Tests, einschließlich Hämogramm, Glukämie, Leber- und Nierenfunktion, Erythrozytensedimentationsrate, Schilddrüsenhormone im Serum, Hyperkoagulabilitäts-Screening, Vaskulitis-Screening, EKG, Röntgenaufnahme des Brustkorbs, Syphilis- und HIV-Serologie und Ultraschall der supraaortalen Trunci mit Doppler, waren normal, mit Ausnahme einer Cholesterinämie von 220 mg/dl mit einem LDL-Cholesterin von 152 mg/dl. Die Hautbiopsie war ebenfalls normal. Im vierten Exon des NOTCH-3-Gens wurde eine Arg169Cys-Mutation (Cytosin-Thymin-Substitution im 505-Codon) gefunden, die als pathogenetisch gilt, so dass die Diagnose CADASIL gestellt wurde. Mit dieser Diagnose wurden beide Eltern in unserer Klinik untersucht.
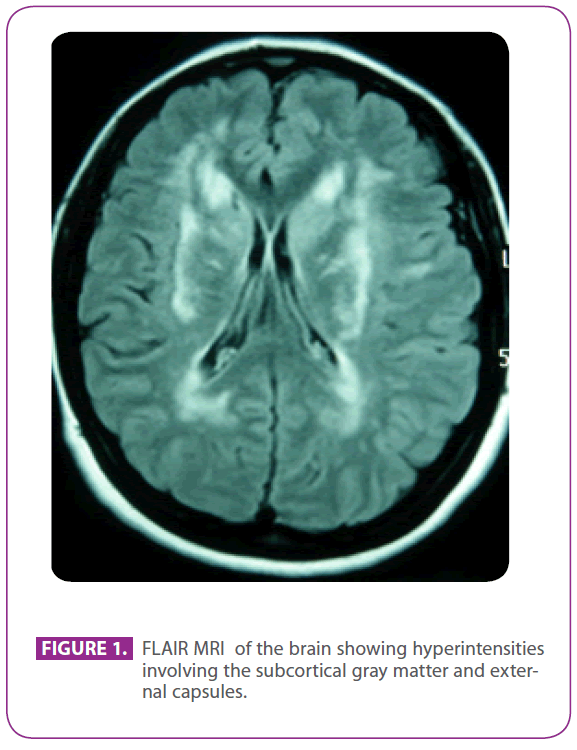
Abbildung 1: FLAIR-MRT des Gehirns mit Hyperintensitäten in der subkortikalen grauen Substanz und den äußeren Kapseln.
Fall 2
Die Mutter war 78 Jahre alt. Sie litt seit langem an Migräne ohne Aura und hatte in den letzten zwei Jahren eine leichte Demenz mit symmetrischem Parkinsonismus, Fluktuationen und REM-Schlaf-Verhaltensstörung entwickelt, die auf eine Demenz mit Lewy-Körperchen hindeutet. Ein MRT des Gehirns schloss eine relevante Erkrankung der weißen Substanz aus.
Ihr Vater war ein 79-jähriger Mann mit mehreren vaskulären Risikofaktoren: Diabetes mellitus Typ 2, Dyslipidämie und Rauchen seit 50 Jahren mit einem kumulierten Index von 15 Packungsjahren, obwohl er vor 3 Jahren damit aufgehört hatte. Er hatte eine ischämische Herzerkrankung mit einem posteroinferioren Myokardinfarkt im Jahr 1991. In den letzten 8 Jahren hatte er drei transitorische ischämische Unfälle und einen lakunären Schlaganfall in verschiedenen Arteriengebieten erlitten, mit fortschreitender Gangstörung, und in den letzten 6 Monaten war er etwas zurückgezogen und apathisch geworden. Er hatte keine Migräne in der Vorgeschichte. Er nahm Metformin, Clopidogrel, Atenolol und Atorvastatin ein. Bei der körperlichen Untersuchung war er bei Bewusstsein und orientiert, aber etwas unaufmerksam, Sprache, Fern- und Kurzzeitgedächtnis waren normal, er zeigte eine ideomotorische Apraxie, eine veränderte Interpretation von Ähnlichkeiten und Sprüchen und keine frontalen Auslösereflexe. Außerdem zeigte er eine globale Hyperreflexie mit bilateralen Flexor-Plantar-Reflexen und einen Gang mit kurzen Schritten und eingeschränkten Armbewegungen. Ein CT-Scan, der zwei Jahre zuvor nach einem seiner transitorischen ischämischen Anfälle durchgeführt worden war, zeigte eine ausgedehnte Erkrankung der weißen Substanz, die besonders in der äußeren Kapsel und den Temporallappen auffiel (Abbildung 2).
Zusammenfassend lässt sich sagen, dass der Patient rezidivierende ischämische Schlaganfälle, eine leichte subkortikale kognitive Beeinträchtigung und eine Leukoenzephalopathie aufwies, was in Anbetracht der Diagnose seiner Tochter höchst verdächtig für CADASIL war. Es wurde ein Gentest durchgeführt, der die gleiche NOTCH-3-Mutation wie bei der Tochter ergab und die Diagnose bestätigte.
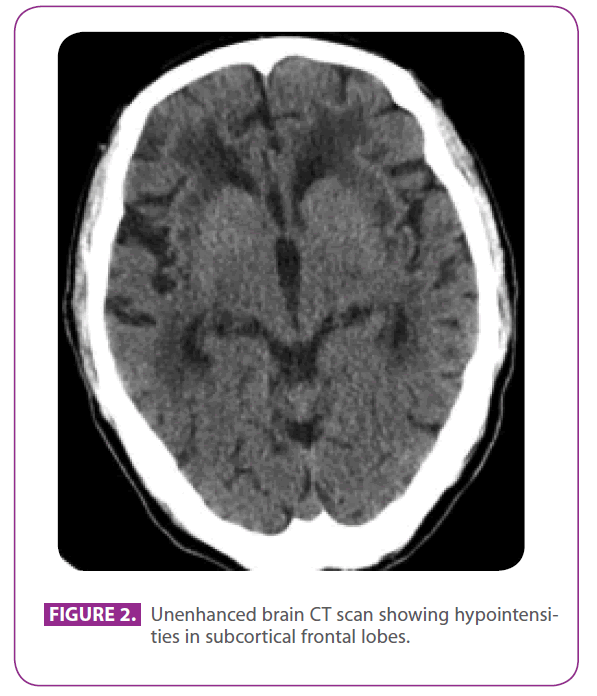
Abbildung 2: Unenhanced brain CT scan showing hypointensities in subcortical frontal lobes.
Discussion
CADASIL ist die häufigste erbliche Erkrankung der kleinen Gefäße. Sie kann aber auch sporadisch auftreten, da De-novo-Mutationen beschrieben worden sind. Sie wird durch verschiedene pathogene Mutationen im NOTCH-3-Gen verursacht, das sich auf Chromosom 19, Locus 19p13.2-p13.1, befindet und aus 33 Exons besteht, die für ein Protein mit 2321 Aminosäuren kodieren (1). Dieses Protein ist ein Single-Pass-Transmembran-Zelloberflächenrezeptor, der in systemischen arteriellen glatten Muskelzellen exprimiert wird und eine extrazelluläre Regulator-Domäne und eine intrazelluläre Transduktor-Domäne besitzt. Bisher wurden mehr als 190 Mutationen gemeldet, die zu CADASIL führen können, und alle treten in den Exons 2 bis 24 des NOTCH-3-Gens auf, die für die 34 epidermalen Wachstumsfaktor-ähnlichen Wiederholungen des extrazellulären Teils von NOTCH 3 kodieren. Daher hat das Screening dieser 23 Exons eine 100 %ige Empfindlichkeit und fast die gleiche Spezifität (2). Von diesen Mutationen sind mehr als 180 Missense-Mutationen, mindestens 6 Deletionen, eine Insertion, ein Frameshift und 2 Duplikationen. Die meisten der pathogenetischen NOTCH3-Mutationen treten in den Exons 3 und 4 auf (3).
Alle Mutationen führen zu einer Hinzufügung oder einem Verlust eines Cysteinrestes in einem EGF-ähnlichen Repeat und damit zu einer ungeraden Anzahl von Cysteinresten, was die Bildung anormaler Disulfidbrücken bewirkt. Die NOTCH3-Mutation verursacht eine Degeneration der glatten Gefäßzellen in kleinen Arterien und Arteriolen und eine Anhäufung des abnormen Proteins in der Wand dieser Gefäße, was zu einer Verengung des Lumens führt (1). Im Gehirn sind die penetrierenden zerebralen und leptomeningealen Arterien betroffen, was zu einer Unfähigkeit dieser Gefäße zur Selbstregulation und zu einer Hypoperfusion der von ihnen durchflossenen Gebiete und damit zu Infarkten in der weißen Substanz führt.
Das erste Symptom von CADASIL ist in der Regel Migräne mit Aura, die im Durchschnitt im Alter von 30 Jahren auftritt. Sie tritt bei 20 bis 40 % der Patienten auf (4). Die meisten Attacken sind typisch mit visueller oder sensorischer Aura, aber die Hälfte der Patienten hat auch atypische Attacken mit basilarer, halbseitiger oder verlängerter Aura.
Subkortikale ischämische Ereignisse, transitorische Attacken oder Schlaganfälle, treten bei 60 bis 85 % der Patienten auf, die erste in einem Durchschnittsalter von 50 Jahren, obwohl sie bereits im zweiten Jahrzehnt auftreten kann. In den meisten Fällen gibt es keine konventionellen vaskulären Risikofaktoren oder sie sind nicht sehr wichtig. Bei zwei Dritteln der Patienten treten die ischämischen Ereignisse klinisch und radiologisch als lakunäre Syndrome auf. Die meisten Patienten erleiden mehrere Schlaganfälle, in der Regel 2 bis 5, die nach einigen Jahren zu Gangstörungen, Harn- und Stuhlinkontinenz, Demenz und Pseudobulbärparese führen (1). Kognitive Beeinträchtigungen sind die zweithäufigste klinische Manifestation. Das früheste Anzeichen ist in der Regel eine Beeinträchtigung der exekutiven Fähigkeiten und der Verarbeitungsgeschwindigkeit, die bei den meisten Patienten im Alter von über 35 Jahren auftritt, aber auch schon im ersten Lebensjahrzehnt auftreten kann (5). Diese kognitive Beeinträchtigung ist progressiv und verschlimmert sich in der Regel bei wiederholten Schlaganfällen, wobei zusätzlich Beeinträchtigungen bei instrumentellen Tätigkeiten, Gedächtnis, Sprache, logischem Denken und visuell-räumlichen Fähigkeiten auftreten. Über 70 % der Patienten sind bis zum sechsten Lebensjahrzehnt dement. Schwere Aphasie, Apraxie oder Agnosie sind bei CADASIL selten.
Psychiatrische Störungen, hauptsächlich Stimmungsstörungen, treten bei 20 % der Patienten auf, im Allgemeinen in Form schwerer depressiver Episoden. Apathie tritt bei 40 % der Patienten auf und ist nicht mit einer Depression verbunden. Andere, weniger häufige klinische Manifestationen sind akute reversible Enzephalopathie (6) (bei 10 % der Patienten), die meist nach einer Migräne mit Aura auftritt, Krampfanfälle (bei 5 bis 10 % der Patienten), Taubheit, Parkinsonismus, Hirnblutungen (meist bei Patienten mit hohem Blutdruck) und Herzinfarkt.
Trotz seiner vollständigen Penetranz weist CADASIL eine erhebliche Variabilität der klinischen Ausprägung zwischen und innerhalb der Familie auf. Die gleiche NOTCH-3-Mutation weist ein breites klinisches Spektrum auf, wobei es keine klaren Unterschiede zwischen homozygoten und heterozygoten Patienten gibt. Tatsächlich gibt es keine Korrelation zwischen Genotyp und klinischem Phänotyp (4). Der Grund für diese Unterschiede ist nicht bekannt, aber es wurden einige mögliche Modifikatoren des Phänotyps von CADASIL beschrieben, wie z. B. aktuelles Rauchen für das Schlaganfallrisiko und das Alter des ersten Schlaganfalls, Bluthochdruck für das Schlaganfallrisiko oder Homocysteinspiegel für das Alter des Auftretens von Migräne (3,4). Diese und andere Faktoren könnten die klinische Ausprägung der Krankheit beeinflussen, indem sie die Genexpression modifizieren oder andere physiopathologische Wege beeinflussen, die zu den gleichen klinischen Manifestationen führen können.
Radiologische Veränderungen treten bei allen Personen mit einer CADASIL-Mutation vor dem 35. Die frühesten und häufigsten Merkmale sind hyperintense, nicht verstärkende punktförmige Areale in der zerebralen weißen Substanz und den subkortikalen Strukturen in T2-gewichteten und flüssigkeitsabgeschwächten Inversionsrecovery-Bildern der MRT. Die Beteiligung der Außenkapseln und des vorderen Teils der Temporallappen ist typisch für diese Erkrankung und ein Anhaltspunkt für die Diagnose, da sie bei der Differenzialdiagnose mit anderen Erkrankungen, wie z. B. der Kleingefäßerkrankung, hilfreich sind (1).
CADASIL wird gemeinhin als Erkrankung junger und mittelalterlicher Erwachsener angesehen, aber die Zahl der älteren Patienten könnte unterdiagnostiziert sein (7,8). Unser zweiter Fall mag als Beispiel dienen: ein 79-jähriger Patient mit vaskulären Risikofaktoren und wiederkehrenden Schlaganfällen, mit einer Erkrankung der weißen Substanz, die von mehreren Neurologen als sekundäre Erkrankung der kleinen Gefäße angesehen wurde. Die Diagnose wurde erst nach Kenntnis der Mutation seiner Tochter gestellt. In Fällen ohne klare Familienanamnese können ein offener Geist und eine sorgfältige Beachtung suggestiver radiologischer Befunde wie die Beteiligung der äußeren Kapsel oder der vorderen Temporallappen der einzige Anhaltspunkt für eine korrekte Diagnose sein.
- Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurol 2009; 8:643-53.
- Vazquez do Campo R, Morales-Vidal S, Randolph C, Chadwick L, Biller J. CADASIL: a case series of 11 patients. Rev Neurol. 2011; 52:202-10.
- Adib-Samii P, Brice G, Martin RJ, Markus HS. Klinisches Spektrum von CADASIL und der Einfluss von kardiovaskulären Risikofaktoren auf den Phänotyp: Studie an 200 konsekutiv rekrutierten Personen. Stroke 2010; 4:630-4.
- Singhal S, Bevan S, Barrick T, Rich P, Markus HS. Der Einfluss von genetischen und kardiovaskulären Risikofaktoren auf den CADASIL-Phänotyp. Brain. 2004; 127:2031-8.
- Dichgans M, Markus HS, Salloway S, Verkkoniemi A, Moline M, Wang Q, Posner H, Chabriat HS. Donepezil bei Patienten mit subkortikaler vaskulärer kognitiver Beeinträchtigung: eine randomisierte Doppelblindstudie in CADASIL. Lancet Neurol. 2008;7:310-8.
- Schon F, Martin RJ, Prevett M, Clough C, Enevoldson TP, Markus HS. „CADASIL coma“: an underdiagnosed acute encephalopathy. J Neurol Neurosurg Psychiatry. 2003;74:249-52.
- Liem MK, Lesnik Oberstein SA, Vollebregt MJ, Middelkoop HA, van der Grond J, Helderman-van den Enden AT. Homozygotie für eine NOTCH3-Mutation bei einem 65-jährigen CADASIL-Patienten mit leichten Symptomen: ein Familienbericht. J Neurol. 2008 ;255:1978-80.
- Lee YC, Yang AH, Soong BW. Die bemerkenswert variable Expressivität von CADASIL: Bericht über einen Mann mit minimalen Symptomen im fortgeschrittenen Alter. J Neurol. 2009;256:1026-7
Schreibe einen Kommentar