Raport przypadku
Clinical Findings
Do 65-letniej kobiety zgłoszono się z licznymi bezobjawowymi dyskretnymi guzkami i blaszkami zanikowymi na udach, utrzymującymi się od 4 lat. Zmiany chorobowe rozpoczęły się jako 2 małe, bezobjawowe, czerwono-szmaragdowe blaszki symetrycznie zlokalizowane na przedniej powierzchni każdego uda, których liczba i wielkość stopniowo rosła, szczególnie na prawym udzie. Dwa lata później na przedniej powierzchni każdego z nich pojawiły się 2 nowe blaszki zanikowe. Zmiany rozwijały się powoli, ale nigdy nie uległy remisji i w kilku poradniach zostały błędnie zdiagnozowane jako pierwotny plamisty zanik skóry. Ogólny stan zdrowia pacjentki był dobry, a jej historia osobista i rodzinna była bez zmian.
Badanie fizykalne ujawniło liczne madder red plaques and nodules of various shapes and sizes (ie, 1-3 cm in diameter) on the anterior aspect of the right thigh. Zmiany były lekko uniesione, o woskowatej powierzchni, twarde i niebolesne przy palpacji. Jedną podobną zmianę zauważono na przedniej powierzchni lewego uda. Dwie 2-centymetrowe, brązowo-czerwone, zanikowe blaszki również zauważono w symetrycznym rozmieszczeniu na przedniej stronie każdego uda. Powierzchnie blaszek były lekko karbowane i błyszczące, a zmiany anetodermalne dawały przy palpacji objaw dziurki od guzika identyczny z nerwiakowłókniakiem (ryc. 1).
 Rycina 1. Na przedniej powierzchni obu ud zaobserwowano wiele czerwonych blaszek, guzków i blaszek zanikowych o różnych kształtach i rozmiarach.
Rycina 1. Na przedniej powierzchni obu ud zaobserwowano wiele czerwonych blaszek, guzków i blaszek zanikowych o różnych kształtach i rozmiarach. Znaleziska histopatologiczne
Dwie próbki biopsyjne zostały pobrane z guzka i blaszki zanikowej na prawym udzie. Badanie mikroskopowe ujawniło odkładanie się jednorodnego materiału eozynofilowego w siateczkowej skórze właściwej i podskórnej, jak również wokół drobnych naczyń (Rycina 2A). W skórze właściwej, zwłaszcza w sąsiedztwie złogów i wokół naczyń, występował łagodny naciek komórkowy złożony z limfocytów, komórek plazmatycznych i komórek olbrzymich (ryc. 2B). Jednorodny materiał miał barwę łososioworóżową przy barwieniu czerwienią Kongo i jasnozieloną przy barwieniu tioflawiną T w mikroskopie fluorescencyjnym (ryc. 3 i 4). Wyniki te sugerowały charakterystyczne cechy skórnej amyloidozy guzkowej.
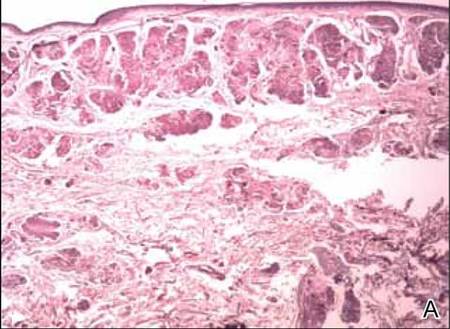
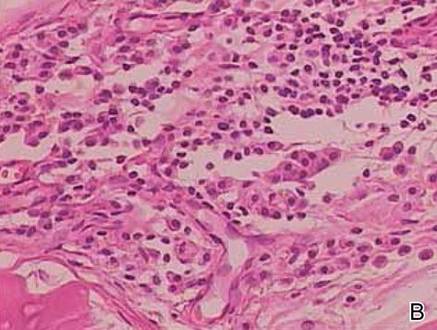
Rycina 2. Histopatologicznie stwierdzono jednorodny materiał eozynofilowy odkładający się w skórze właściwej siateczkowatej i podskórnej (A)(H&E, oryginalne powiększenie ×25). W badaniu mikroskopowym stwierdzono nacieki z limfocytów i komórek plazmatycznych w skórze właściwej, zwłaszcza w sąsiedztwie złogów i wokół naczyń (B)(H&E, oryginalne powiększenie ×200).
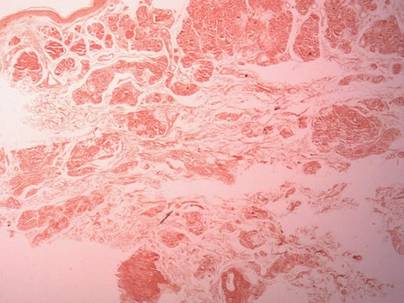
Rycina 3. Barwienie czerwienią Kongo wykazało łososioworóżowy jednorodny materiał (oryginalne powiększenie ×25).
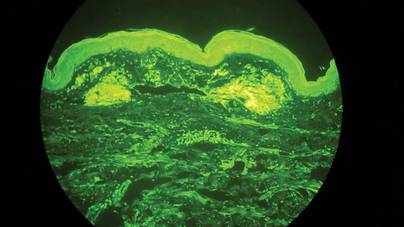
Rycina 4. Thioflavin T staining showed bright green homogeneous material (original magnification ×100).
Laboratory Findings
Laboratory studies showed normal results for complete blood cell count, urinalysis, liver and renal function tests, blood glucose levels, lipid panel, and erythrocyte sedimentation rate. Elektroforeza białek surowicy była prawidłowa i nie wykryto białek Bence Jonesa. Poziomy IgA, IgG i IgM w surowicy nie wykazały nieprawidłowości. Elektrokardiogram, radiografia klatki piersiowej i ultrasonografia jamy brzusznej były prawidłowe.
Na podstawie wyników klinicznych, histopatologicznych i laboratoryjnych postawiono rozpoznanie pierwotnie zlokalizowanej skórnej amyloidozy guzkowej (PLCNA). Mimo że zaproponowano etapowe wycięcie chirurgiczne, pacjentka odmówiła leczenia, ponieważ zmiany były bezobjawowe. Podczas 2,5-letniej obserwacji nie stwierdzono wyraźnej progresji zmian skórnych ani nieprawidłowych wyników badań ogólnoustrojowych.
Komentarz
Amyloidoza to spektrum chorób polegających na odkładaniu się białek amyloidowych w różnych tkankach. Pod względem klinicznym amyloidozę dzieli się na pierwotne i wtórne postacie amyloidozy układowej, amyloidozę związaną z hemodializą, amyloidozę rodzinną oraz amyloidozę skórną. Pierwotna amyloidoza skórna jest zlokalizowana na skórze bez zajęcia innych narządów i nie występuje w amyloidozie układowej. Wtórne zajęcie skóry w amyloidozie układowej jest rzadkie. Większość przypadków pierwotnie zlokalizowanej skórnej amyloidozy (PLCA) jest sporadyczna, ale około 10% przypadków może występować rodzinnie.1 Wyróżnia się 3 główne formy zlokalizowanej skórnej amyloidozy: amyloidozę plamistą, liszajowatą i guzkową. Amyloidoza guzkowa jest najrzadszą postacią PLCA.
Amyloidoza guzkowa została po raz pierwszy opisana przez Gottrona w 1950 r.2 Zmiany skórne mogą występować jako pojedyncze lub mnogie guzki, czasami z nakładającymi się blaszkami zanikowymi. Zmiany składają się z twardych, gładkich, woskowatych lub gumowatych, różowych do opalonych grudek, blaszek lub guzków o wielkości do kilku centymetrów. Na niektórych zmianach mogą być widoczne powierzchniowe teleangiektazje. Opisywano zmiany o wyglądzie pęcherzykowatym i anetodermalnym.3 Najczęstszą lokalizacją jest okolica tarczowa, a następnie odpowiednio nogi, głowa, tułów, ramiona i genitalia.4 W niektórych przypadkach zmiany mogą samoistnie ustępować z upływem czasu. U naszego pacjenta zmiany składały się zarówno z licznych guzków, jak i blaszek zanikowych, co jest rzadkością.
Patogeneza odkładania się amyloidu jest nadal nieznana. Skórna amyloidoza plamista i liszajowata może pochodzić ze zdegenerowanych filamentów pośrednich keratynocytów. Amyloidoza guzkowa może reprezentować zlokalizowaną dyscrazję komórek plazmatycznych, która może być związana z gammopatią monoklonalną lub szpiczakiem mnogim.5 Niektóre składniki amyloidu w niektórych przypadkach PLCNA mogą składać się z łańcuchów lekkich immunoglobulin κ i λ, przy czym większość zgłoszonych przypadków dotyczy podtypu l.6 Wyniki jednego z badań wskazały, że β2-mikroglobulina była kolejnym głównym składnikiem fibryli amyloidowych i że β2-mikroglobulina była częściowo poddawana modyfikacji zaawansowanego końcowego produktu glikacji w PLCNA.7
.
Dodaj komentarz