Introduction
Projektowanie i wytwarzanie nanokompozytów glina/polimer wzbudziły duże zainteresowanie zarówno społeczności naukowej, jak i inżynierskiej (Cheng et al., 2017). Opracowano szeroką gamę nanokompozytów glina/polimer, które znajdują szerokie zastosowanie (Gogoi i Raidongia, 2017). Wstępne interkalowanie glin, a następnie inkorporacja polimerów do warstwy minerałów ilastych jest szeroko przyjętym podejściem do przygotowania nanokompozytów glina/polimer (Li et al., 2009; Zare et al., 2017). Podczas interkalacji w obrębie nieorganicznych warstw gliny, łańcuchy polimerów organicznych w naturalny sposób zmniejszają swoją mobilność strukturalną, a niektóre z nich przyjmują wysoce zorganizowaną konformację w obrębie struktury warstwowej.
W naturze kaolinit (Kaol) jest rodzajem minerału ilastego o strukturze warstwowej krzemianu glinu dioktaedrycznego 1:1 (Brindley i Robinson, 1945). Warstwy w Kaol są utrzymywane razem przez wiązania wodorowe, oddziaływania dipol-dipol i siły van der Waalsa (Brindley et al., 1967). Jednakże, tylko ograniczona liczba wysoce polarnych gatunków organicznych, w tym mocznik (Makó i in., 2009), sulfotlenek dimetylu (Costanzo i Giesse, 1986), formamid (Frost i in., 2000a), hydrazyna (Cruz i Franco, 2000) i octan potasu (Frost i in., 2000b) zostały z powodzeniem interkalowane do galerii Kaolu. Interkalacja małych cząsteczek w warstwach Kaolu powoduje zwiększenie odstępu bazowego i może być wykorzystana jako wstępny etap ekspansji dla późniejszego wprowadzania dużych rozmiarów, niereaktywnych gatunków poprzez przemieszczenie wstępnie interkalowanych małych cząsteczek (Cheng i in., 2015). Na przykład, na podstawie wcześniejszych doniesień, związek interkalacyjny Kaol/metanol może być efektywnym półproduktem do dalszej reakcji interkalacji z glikolem etylenowym (Hirsemann i in., 2011), heksyloaminą (Matusik i in., 2012), n-alkiloaminami (Gardolinski i Lagaly, 2005) oraz czwartorzędowymi solami amoniowymi (Cheng i in., 2016). Ponadto większość cząsteczek gości jest trudna do wprowadzenia w przestrzeń międzywarstwową, jednym z powodów są silne wiązania wodorowe, innym brak jonów wymiennych w strukturze Kaol.
Although clay/polymer nanocomposites have been well developed in recent years, to to synthesize new clay/organic nanocomposites for potential application still presents a big challenge (Kotal and Bhowmick, 2015). W celu rozwiązania tego problemu, naprawdę pilną sprawą jest poznanie procesu interkalacji kaolinitu w skali nano oraz przeprowadzenie dokładnych badań mechanizmu rozkładu przy użyciu nowoczesnych technologii i narzędzi analitycznych. Analiza termiczna i obliczenia kinetyczne związków interkalacyjnych glina/polimer mogą pomóc w scharakteryzowaniu procesów rozkładu i zapewnić podstawy naukowe do kontroli reakcji interkalacyjnych (Zhang et al., 2015). Parametry kinetyczne procesu kinetyki reakcji rozkładu, takie jak energia aktywacji, pre-eksponencjalna, rzędy reakcji i stała szybkości, zostały ocenione na podstawie danych z termogramów gliny/polimeru. W niniejszej pracy zbadano interkalację Kaolu różnymi alkiloaminami. Dodatkowo, systematycznie badano proces interkalacji i mechanizm rozkładu.
Doświadczalne
Materiały
Kaolin użyty w tej pracy został pozyskany z Zhangjiakou, Chiny. Głównym składem mineralnym jest dobrze uporządkowany kaolinit (95% masy). Przed interkalacją zmielono go tak, aby przeszedł przez sito o oczkach 325 (cząstki o wymiarach < 44 μm). Heksyloaminę alkilową (HA, 99%), dodecyloaminę (DA, chemicznie czysta) i oktadecyloaminę (OA, chemicznie czysta) zakupiono w Nanjing Shuguang Chemical Company, Chiny. Dimetylosulfotlenek (DMSO, odczynnik analityczny), metanol (MeOH, odczynnik analityczny) i toluen (99%) otrzymano z Xilong Chemical Company, Chiny, i stosowano w postaci, w jakiej je otrzymano, bez dalszego oczyszczania. Wzór chemiczny i wzór strukturalny HA, DA i OA przedstawiono w Tabeli 1.

Tabela 1. Wzór chemiczny i strukturalny HA, DA, i OA.
Synteza związków interkalacyjnych
Pierwszy, związek interkalacyjny Kaol/DMSO został przygotowany przez zdyspergowanie 20,0 g Kaolu w mieszaninie 36,0 g DMSO i 4,0 g wody. Mieszaninę mieszano w łaźni wodnej przez 2 h w temp. 95°C, a następnie zawiesinę rozdzielono przez odwirowanie z etanolem. Po drugie, związek interkalacyjny Kaol/DMSO wykorzystano jako prekursor do dalszej reakcji z MeOH. Do interkalowanego Kaolu dodano MeOH, a mieszaninę reakcyjną mieszano przez 10 dni, przy czym MeOH zastępowano codziennie taką samą ilością świeżego MeOH. Osad w mieszaninie oddzielono przez odwirowanie i suszono w piecu w temperaturze 60°C przez 12 h, otrzymując związki interkalacyjne Kaol/MeOH. Na koniec, 2 g związku interkalacyjnego Kaol/MeOH mieszano z 30.0 mL roztworów metanolowych HA, DA, lub OA (1 mol/L) mieszając odpowiednio w temperaturze otoczenia. Dyspersje wirowano po reakcji przez 24 h. Osady przemywano trzykrotnie toluenem w celu usunięcia nadmiaru HA, DA lub OA. Próbki suszono w temperaturze pokojowej przez 12 h i rozdrabniano na proszki za pomocą moździerza agatowego (Komori i in., 1999). Otrzymane związki oznaczono jako Kaolpi/HA (pi jako wstępnie interkalowane), Kaolpi/DA, oraz Kaolpi/OA.
Charakterystyka
Wzory XRD rejestrowano na dyfraktometrze rentgenowskim Rigaku D/max 2500PC z promieniowaniem Cu Kα (λ = 1.54178 Å) pracującym przy napięciu 40 kV i natężeniu 150 mA. Losowo zorientowane próbki były skanowane w zakresie 2θ od 1 do 20° z prędkością 2° min-1. Spektrofotometr Thermo Fisher Nicolet 6700 służył do rejestracji widm FTIR w zakresie 4000~400 cm-1. Analizę TG-DSC przeprowadzono na analizatorze termicznym Mettler-Toledo TG-DSC I/1600 HT w atmosferze azotu. Dwadzieścia miligramów próbki umieszczono w tyglu z tlenku glinu i ogrzewano od 30 do 1100°C z serią szybkości ogrzewania 4, 6, 8, 10°C min-1.
Wyniki i dyskusja
Charakterystyka XRD
Wzory XRD dziewiczego Kaolu i jego związków interkalacyjnych przedstawiono na rysunku 1. Wzór czystego Kaol wyświetla dobrze uporządkowaną strukturę warstwową z odstępem bazowym 0,71 nm. Wartość ta dobrze pasuje do standardowego wzorca odniesienia ICDD 14-0164 . Po potraktowaniu DMSO, nowy bazowy pik odbicia pojawił się przy 1,14 nm, wskazując, że DMSO został pomyślnie wprowadzony do przekładek Kaol. Po poddaniu związku interkalacyjnego Kaol/DMSO działaniu MeOH, pik odbicia przy 1,14 nm (001) charakterystyczny dla związku interkalacyjnego Kaol/DMSO został przesunięty do 0,86 nm. Matusik et al. (2012) podali, że związek Kaol/DMSO/MeOH został wysuszony w temperaturze 110°C, odbicie o d = 1,12 nm zniknęło i zaobserwowano szerokie odbicie z maksimum przy 0,95 nm. W oparciu o wzór chemiczny Al2Si2O5(OH)3.20(OCH3)0.80 związku interkalacyjnego Kaol/DMSO, który został obliczony na podstawie analizy CHNS, wykazano, że około 1/3 wewnętrznych powierzchniowych grup OH została zastąpiona grupami metoksy, a obserwacja ta również sugeruje tworzenie się związku interkalacyjnego Kaol/MeOH (Tunney i Detellier, 1996; Cheng i in., 2015). Wzorce XRD pokazują również, że związki Kaol/MeOH interkalowane z HA, DA i OA rozszerza strukturę wzdłuż osi c, co prowadzi do dużej odległości międzywarstwowej wynoszącej odpowiednio 2,86, 4,08 i 5,66 nm. Długości łańcuchów molekularnych HA, DA i OA wynoszą odpowiednio 1,56, 2,51 i 3,65 nm (McNulty i in., 2011, 2014). Ogólnie, odległość międzywarstwowa związków interkalacyjnych Kaol/alkiloaminy zwiększała się wraz z długością łańcucha alkilowego alkiloamin, ale cząsteczka alkiloaminy nie jest strukturą jednowarstwową lub dwuwarstwową, która jest prostopadła do powierzchni Kaol.
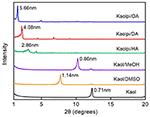
Figura 1. Wzory XRD dla Kaol, Kaol/DMSO, Kaol/MeOH, Kaolpi/HA, Kaolpi/DA, i Kaolpi/OA.
Widma FTIR
Spektroskopia FTIR była szeroko stosowana w charakteryzacji związków interkalacyjnych (Ledoux i White, 1964; Frost et al., 2000c; Cheng et al., 2012). Widma FTIR oryginalnego Kaol, Kaolpi/HA, Kaolpi/DA oraz związków interkalacyjnych Kaolpi/OA przedstawiono na rysunku 2. Charakterystyczne pasma przy 432, 470 i 541 cm-1 (rysunek 2B) należą do trybu deformacji Si-O, Si-O-Si i Al-O-Si. Pozostałe pasma przy 1,009, 1,031 i 1,114 cm-1 (rysunek 2C) są przypisane do drgań rozciągających Si-O-Si w warstwie Kaol. Porównując widma Kaolu i jego związków interkalacyjnych, położenie pasm nie wykazuje wyraźnych zmian, co wskazuje, że na te pasma drgań rozciągających nie mają wpływu interkalowane cząsteczki.
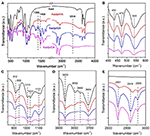
Rysunek 2. Widma w podczerwieni Kaol, Kaolpi/HA, Kaolpi/DA, Kaolpi/OA.
Paski przy 912 i 938 cm-1 (Rysunek 2C) należą do deformacji OH wewnętrznych grup hydroksylowych i wewnętrznych-powierzchniowych grup hydroksylowych. Istnieją cztery oczywiste pasma rozciągania hydroksylu na 3,694, 3,669, 3,652 i 3,620 cm-1 (rysunek 2D) w widmie oryginalnego Kaol, pasma na 3,694, 3,669 i 3,652 cm-1 są spowodowane przez wewnętrzne-powierzchniowych grup hydroksylowych, a jeden na 3,620 cm-1 wynika z wewnętrznych grup hydroksylowych (Farmer, 1964; Ledoux i White, 1964; Giese i Datta, 1973). Po interkalacji cząsteczek alkiloaminy niektóre pasma uległy osłabieniu na skutek zmiany oddziaływań z sąsiednimi atomami. Przesunięcie do 3,697 cm-1 i zmniejszenie intensywności pasma przy 3,694 cm-1 wskazuje, że liczne grupy hydroksylowe w Kaolu uczestniczyły i tworzyły wiązania wodorowe z grupami aminowymi w cząsteczkach alkiloamin (Caglar, 2012; Caglar i in., 2013; Zhang i in., 2015).
Obecność międzywarstwowych cząsteczek alkiloamin wykryto na widmie FTIR poprzez nowe piki, które przedstawiono na rysunkach 2A,E. Dla związków interkalacyjnych, niektóre charakterystyczne piki, takie jak pasmo przy 1,388 i 1,468 cm-1 należą do trybu zginania C-H odpowiednio -CH3 i -CH2. Zaobserwowano nowe pasmo drgań przy 3,334 cm-1 , które jest spowodowane trybem rozciągającym drgań rozciągających N-H (Griffiths i De Haseth, 2007; Zhang et al., 2018). Zaobserwowano nowe pasmo drgań przy 2,956 cm-1, które zwykle przypisuje się do pasma asymetrycznego rozciągania C-H terminalnych grup metylowych (Cheng i in., 2016). Dwa pasma drgań przy 2,919 i 2,851 cm-1 są zwykle przypisywane symetrycznym i asymetrycznym drganiom rozciągającym -(CH2)n- (Venkataraman i Vasudevan, 2001). Intensywność tych pasm stopniowo wzrasta, co oznacza, że więcej atomów węgla alkiloaminy zostało wbudowanych w warstwy Kaolu. Ponadto, lotność alkiloamin zmniejsza się wraz ze wzrostem łańcucha węglowego, więc stabilność odpowiednich związków interkalacyjnych Kaol będzie stopniowo wzrastać wraz z większą masą cząsteczkową alkiloaminy (Wang et al., 2010).
Structural Model of Kaol/alkylamine Intercalation Compounds
Układ interkalowanych cząsteczek jest podstawą do eksperymentalnej analizy struktury związków interkalacyjnych Kaol/alkiloamina. Według Lagaly’ego (1981), modele strukturalne związków interkalacyjnych montmorillonit-quaternary ammonium salt można podzielić na trzy kategorie: (a) monowarstwy: krótkołańcuchowe jony alkiloaminowe; (b) bilayery: długołańcuchowe czwartorzędowe jony amoniowe; (c) trójwarstwy: zagięte łańcuchy alkilowe w silnie naładowanych minerałach ilastych. Beneke i Lagaly (1982) na podstawie badań XRD zaproponowali różne struktury interkalowanych cząsteczek alkiloamin, które zostały podsumowane następująco: (a) łańcuchy alkilowe w przestrzeni międzywarstwowej są nachylone do płaszczyzny krystalicznej (001); (b) łańcuchy alkilowe są ułożone w bilayery. Brindley i Moll (1965) opisali, że interkalowane molekuły są nachylone do arkuszy krzemianowych w pojedynczej warstwie, podczas gdy kąt wynosi około 65° do płaszczyzny krystalicznej (001). Jeśli oddziałujące cząsteczki są jednakowo przyłączone do wszystkich powierzchni krzemianowych przez aktywne grupy hydroksylowe, sugeruje się organizację „koniec do końca” w parach z pewnym przemieszczeniem wzdłużnym. Kąt nachylenia jest zgodny z możliwym sposobem ścisłego upakowania pomiędzy powierzchniami tlenowymi krzemianów i końcowymi grupami hydroksylowymi oraz stopniem ścisłego upakowania cząsteczek łańcucha między sobą.
Cząsteczki alkilowe są trudne do interkalacji bezpośrednio do oryginalnych warstw Kaol, ponieważ brakuje jonów wymiennych w przestrzeni międzywarstwowej Kaol. Ważne prekursory są niezbędne, gdy cząsteczki alkiloaminy wstawić do międzywarstwy Kaol. Donoszono, że łańcuch alkilotrimetylochlorku amonu tworzy w międzywarstwie Kaol przechyloną dwuwarstwę, a kąt przechylenia wynosi po obliczeniach ~36° (Kuroda i in., 2011). Yuan i wsp. (2013) wskazali, że układ strukturalny cząsteczek czwartorzędowej soli amoniowej w międzywarstwie Kaol tilted bilayer i kąt nachylenia wynosi ~38,4°. Gardolinski i Lagaly (2005) zaproponowali, że łańcuchy alkilowe w międzywarstwach są w pełni rozciągnięte i zorientowane prostopadle do powierzchni Kaol przez bilayer. Dlatego też, analizując długość łańcucha cząsteczek HA, DA i OA, można wyciągnąć wniosek, że odstęp bazowy po interkalacji nie jest wystarczający, aby umożliwić łańcuchom alkilowym wejście w sposób pionowy i dwuwarstwowy. Analizując dalej wyniki XRD można zauważyć, że łańcuchy alkilowe HA, DA i OA są nachylone do powierzchni Kaol w dwuwarstwie, a kąt nachylenia łańcuchów alkilowych wynosi odpowiednio około 39,9, 39,9, 41,1°. Ponadto, ze względu na wiązanie hydroksylowe pomiędzy hydroksylem wewnątrzpowierzchniowym w Kaol i grupami aminowymi w cząsteczkach alkiloaminy, najbardziej możliwy model strukturalny dla związków interkalacyjnych Kaol/alkiloamina jest przedstawiony na Rysunku 3.
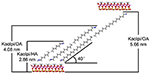
Rysunek 3. Najbardziej możliwy model strukturalny dla związków interkalacyjnych kaolinit/alkiloamina.
Analiza termiczna
Wyniki analizy TG-DSC Kaolu i jego związków interkalacyjnych przedstawiono na rysunku 4. Główny ubytek masy kaolu występuje w temperaturze pomiędzy 400 a 600°C, z maksymalnym ubytkiem w 525°C, przypisywanym utracie wody z powodu dehydroksylacji sieci krystalicznej, co prowadzi do powstania meta-kaolinitu (Toussaint et al., 1963; Criado et al., 1984; Sperinck et al., 2011). Ubytek ten odpowiada około 12,7% całkowitej masy, co jest bardzo bliskie teoretycznej wartości referencyjnej (13,9%). Porównując krzywe TG-DSC oryginalnego Kaolu, w przypadku związków interkalacyjnych Kaolpi/HA, Kaolpi/DA i Kaolpi/OA obserwuje się dwa główne etapy utraty masy. Pierwszy z nich wystąpił pomiędzy 150 a 350°C, co jest spowodowane rozpadem interkalowanych cząsteczek alkiloaminy (Yuan i in., 2013). Drugi, który wystąpił w temperaturze około 500°C jest spowodowany dehydroksylacją deinterkalowanego Kaolu. W przypadku Kaolpi/OA, niektóre OA zostały interkalowane do przestrzeni międzywarstwowej krzemianu, tworząc stabilną interkalację, co może być udowodnione przez wzór XRD. Niektóre cząsteczki są uwikłane wewnątrz siebie z powodu długiego łańcucha lub zacisku w nanorolkach, których nie można w pełni usunąć (Li i in., 2015; Liu i in., 2016). W temperaturze 220°C wyraźny ubytek masy wynika z rozkładu pokrytego lub niezwiązanego OA, natomiast ubytek masy w temperaturze 330°C przypisuje się rozkładowi interkalowanego OA. Pik egzotermiczny w temperaturze 996°C jest wynikiem rekrystalizacji Kaolu.
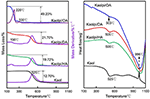
Rysunek 4. Krzywe TG-DSC dla Kaol, Kaolpi/HA, Kaolpi/DA, Kaolpi/OA.
Porównując krzywe TG-DSC oryginalnego Kaol i związków interkalacyjnych Kaol/alkiloamina (Kaolpi/HA, Kaolpi/DA, Kaolpi/OA) można zaobserwować: (a) ma różne etapy ubytku masy, jak również znaczną różnicę w ubytkach masy pomiędzy Kaolpi/OA a pozostałymi dwoma związkami interkalacyjnymi (Kaolpi/HA, Kaolpi/DA). Ubytek masy przy rozkładzie Kaolpi/HA i Kaolpi/DA wynosił około 20%, natomiast w przypadku Kaolpi/OA wynosi on około 50%. Może to być spowodowane tym, że większa ilość OA została interkalowana w przestrzeni międzywarstwowej krzemianu niż HA i DA (Wang et al., 2017). (b) Temperatura dehydroksylacji związków interkalacyjnych była niższa od temperatury oryginalnego Kaol o około 20°C. Wynika to z faktu, że cząsteczki interkalacyjne rozszerzały odstępy międzywarstwowe, osłabiały wiązania wodorowe między warstwami Kaol, doprowadzając do łatwiejszej dehydroksylacji z powierzchni Kaol. W związku z tym, krystaliczność Kaolu gwałtownie spadła po interkalacji, co można potwierdzić metodami XRD i IR (poniżej 800 cm-1). Wynik ten jest zgodny z wnioskiem z niektórych innych doniesień, że temperatura dehydroksylacji była determinowana przez krystaliczność Kaolu (Yeskis i in., 1985; Gabor i in., 1995; Sahnoune i in., 2012).
Kinetyka reakcji rozkładu
Kinetykę degradacji interkalacyjnego związku Kaol/alkiloamina zbadano za pomocą technik termograwimetrii. Parametry kinetyczne (energię aktywacji E i współczynnik pre-eksponencjalny A) procesu degradacji obliczono na podstawie metod KAS i Ozawy (Kissinger, 1957; Škvára i Šesták, 1975). Mechanizm degradacji termicznej związku interkalacyjnego Kaol/alkiloamina badano metodą całek Satava (Xie i in., 2001; Gao i in., 2005; Zhang i in., 2015).
Zgodnie z teorią reakcji, wzór kinetyczny rozkładu nieizotermicznego jest zwykle wyrażony w następujący sposób:
gdzie α jest stopniem konwersji B(s) w czasie t, f (α) jest funkcją mechanizmu reakcji, a k jest stałą szybkości reakcji. k spełnia następujące równanie:
Jeżeli temperatura próbki jest kontrolowana ze stałą szybkością ogrzewania (β = dT/dt), szybkość reakcji można określić następująco:
Po rozdzieleniu zmiennej, ponownym uporządkowaniu za pomocą całek lub funkcji różniczkowych równania (4), równanie KAS (5) i równanie Ozawy (6) można obliczyć w następujący sposób:
Gdzie G(α) reprezentuje integralną funkcję przekształcenia. Ze względu na aproksymację całkową, do obliczenia E zastosowano metody iteracyjne, aby uniknąć pewnych odchyleń. Funkcje metod iteracyjnych są następujące:
Ustawiamy x = E/RT, definicje h(x) i H(x) są następujące:
Iteracja przebiega w następujących trzech krokach:
(a) Ustawić h(x) = 1 lub H(x) = 1, obliczyć E0 metodą najmniejszych kwadratów zgodnie z nachyleniem zależności liniowej lnβ/T2 i lnβ do 1/T.
(c) Zastąpić E0 przez E1, powtarzać krok (b) aż do uzyskania Ei-Ei-1 < 0. 1 kJ mol-1. Ei jest dokładną wartością energii aktywacji reakcji rozkładu.
Wartości E związków interkalacyjnych Kaol/alkiloamina obliczono metodami KAS, Ozawy i iteracyjną (Tabela 2). Wyniki obliczeń uzyskane metodą KAS były bliższe wynikom z metod iteracyjnych, jednak wyniki z metody iteracyjnej Ozawy wykazywały większe odchylenia od pozostałych. Dlatego też średnie E z metod iteracyjnych można uznać za energię aktywacji reakcji rozkładu, gdyż są one lepsze od metod KAS i Ozawy. Średnie E dla Kaolpi/HA, Kaolpi/DA i Kaolpi/OA wynoszą 104,44, 130,80 i 154,59 kJ mol-1. Można zauważyć, że średnie E związków Kaolpi/alkiloamina są dodatnio skorelowane z długością łańcucha alkilowego. Jednym z powodów jest to, że lotność słabnie, jak łańcuch alkilowy rośnie. Innym powodem jest to, że Kaolpi/OA posiada najlepiej uporządkowaną strukturę interkalacyjną, która jest najtrudniejsza do de-interkalacji. A wyniki te mogą być potwierdzone przez wzory XRD.
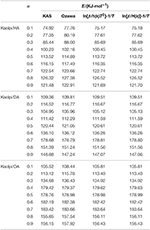
Tabela 2. E związków interkalacyjnych Kaol/alkiloamina o różnej α obliczone metodami KAS, Ozawy i iteracyjną.
Funkcje i A mechanizmu reakcji rozkładu obliczono metodami Satavy, które opisano następująco:
Zastąpić α w każdej funkcji mechanizmu G(α), aby uzyskać wartość lg, dopasowanie liniowe lg i 1/T, które odpowiadają szybkości ogrzewania, funkcja, która ma największe dopasowanie może być określona jako najbardziej prawdopodobna funkcja mechanizmu. A wartości E i A można uzyskać poprzez nachylenie i przechwyt krzywej dopasowania liniowego.
Jak pokazano w tabeli 3, pięć funkcji mechanizmu z dobrocią dopasowania powyżej 90% są wybierane w oparciu o wzór całkowy Satava. Porównując te pięć funkcji, funkcję mechanizmu nr 9 G(α) = 2 o dobroci dopasowania około 97% można uznać za najbardziej prawdopodobną funkcję mechanizmu, a krzywe dopasowania liniowego funkcji nr 9 przedstawiono na rysunku 5. E i A znajdują się również w normalnym zakresie kinetyki rozkładu. Funkcje mechanizmu rozkładu dla trzech związków interkalacyjnych Kaol/alkiloamina są podane w Tabeli 4.

Tabela 3. Wybrane funkcje mechanizmu kinetycznego ocenione metodą całek Satava.

Rysunek 5. Krzywe dopasowania liniowego funkcji nr 9 ocenianej metodą całek Satava.

Tabela 4. Funkcje mechanizmu kinetycznego związków interkalacyjnych Kaol/alkiloamina.
Wnioski
Przy użyciu metod XRD, FT-IR i TG-DSC zbadano możliwe modele struktury i kinetykę rozkładu związków interkalacyjnych Kaol/alkiloamina. Interkalacja związku Kaol/MeOH z HA, DA i OA rozszerza odstępy międzywarstwowe Kaolu wzdłuż osi c, powodując pojawienie się dominującego odbicia odpowiednio przy 2,86, 4,08 i 5,66 nm. Ponadto, odstęp bazowy po interkalacji nie jest wystarczający, aby umożliwić wnikanie łańcuchów alkilowych w sposób pionowy jedno- i dwuwarstwowy. Stwierdzono, że łańcuchy alkinowe HA, DA i OA są nachylone do powierzchni Kaol w dwuwarstwie, a kąt nachylenia łańcuchów alkinowych wynosi ~40°.
Na podstawie metod KAS, Ozawy i iteracyjnej obliczono wyniki energii aktywacji E związków interkalacyjnych Kaol/alkiloamina. Średnia energia aktywacji E związków Kaolpi/HA, Kaolpi/DA i Kaolpi/OA wynosi 104,44, 130,80 i 154,59 kJ mol-1. Średnia energia aktywacji E związków Kaolpi/alkiloamina wykazuje dodatnią korelację z długością łańcucha alkilowego. Zoptymalizowana funkcja mechanizmu procesu rozkładu Kaol/alkiloamina została określona jako dyfuzja 3D z funkcją całkującą G(α) = 2. Badania te są korzystne dla lepszego zrozumienia mechanizmu rozkładu nanokompozytów glinka/organ oprócz dostarczenia inspiracji do syntezy nowych materiałów na bazie glinki.
Wkład Autorów
HC i QfL zaprojektowali eksperyment i zrewidowali pracę. YZ, QhL i PX przeprowadzili eksperymenty i napisali pracę. YZ i QhL w równym stopniu przyczynili się do powstania tej pracy.
Oświadczenie o konflikcie interesów
Autorzy oświadczają, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych powiązań, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Dr Luyi Sun (Department of Chemical and Biomolecular Engineering and Polymer Program, Institute of Materials Science, University of Connecticut) dziękuje za korektę pracy. The authors gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (41602171), the Beijing Natural Science Foundation (8164062) and the Yue Qi Young Scholar Project, China University of Mining & Technology, Beijing.
Giese, R., and Datta, P. (1973). Hydroxyl orientation in kaolinite, dickite, and nacrite. Am. Mineral. 58, 471-479.
Google Scholar
Griffiths, P. R., and De Haseth, J. A. (2007). Fourier Transform Infrared Spectrometry. New York, NY: John Wiley & Sons.
Google Scholar
Yeskis, D., Van Groos, A. K., and Guggenheim, S. (1985). Dehydroxylation of kaolinite. Am. Mineral. 70, 1-2.
Google Scholar
.
Dodaj komentarz