Tapauskertomus
Kliiniset löydökset
65-vuotiaalla naisella todettiin 4 vuotta kestäneitä, useampia oireettomia, erillisiä kyhmyjä ja atrofisia plakkeja reisissä. Vauriot olivat alkaneet kahdesta pienestä, oireettomasta, madderinpunaisesta plakista, jotka sijaitsivat symmetrisesti kummankin reiden etupuolella ja joiden määrä ja koko olivat vähitellen kasvaneet, erityisesti oikeassa reidessä. Kaksi vuotta myöhemmin molempien reisien etupuolelle ilmestyi kaksi uutta atrofista plakkia. Vauriot kehittyivät hitaasti, mutta ne eivät koskaan korjaantuneet, ja useat poliklinikat olivat diagnosoineet ne virheellisesti ihon primaariseksi makulaariseksi atrofiaksi. Potilaan yleisterveys oli hyvä, ja hänen henkilö- ja perhehistoriansa oli merkityksetön.
Kuntotutkimuksessa paljastui useita madderinpunaisia plakkeja ja kyhmyjä, jotka olivat erimuotoisia ja -kokoisia (eli halkaisijaltaan 1-3 cm) oikean reiden etupuolella. Vauriot olivat hieman koholla ja vahamaisen pinnan omaavia, kiinteitä ja kivuttomia tunnusteltaessa. Yksi samanlainen vaurio todettiin vasemman reiden etupuolella. Kummankin reiden etupuolella todettiin myös kaksi 2 cm:n pituista, ruskeanpunaista, atrofista plakkia symmetrisesti jakautuneena. Plakkien pinnat olivat hieman ryppyisiä ja kiiltäviä, ja anetodermia muistuttavat leesiot tuottivat tunnusteltaessa neurofibrooman kanssa identtisen napinläpimerkin (kuva 1).
 kuva 1. Molempien reisien etupuolella nähtiin useita eri muotoisia ja kokoisia madderinpunaisia plakkeja, kyhmyjä ja atrofisia plakkeja.
kuva 1. Molempien reisien etupuolella nähtiin useita eri muotoisia ja kokoisia madderinpunaisia plakkeja, kyhmyjä ja atrofisia plakkeja. Histopatologiset löydökset
Oikean reiden kyhmystä ja atrofisesta plakista otettiin kaksi biopsianäytettä. Mikroskooppinen tutkimus paljasti homogeenisen eosinofiilisen aineen kerrostumisen verkkomaisessa dermiksessä ja subcutiksessa sekä hienojen verisuonten ympärillä (kuva 2A). Lymfosyyttien, plasmasolujen ja jättiläissolujen lievää soluvirtausta esiintyi dermiksessä, erityisesti laskeumien vieressä ja verisuonten ympärillä (kuva 2B). Homogeeninen materiaali näytti lohenpunaiselta kongopunavärjäyksessä ja kirkkaan vihreältä tioflaviini T -värjäyksessä fluoresoivalla mikroskoopilla (kuvat 3 ja 4). Nämä tulokset viittasivat ihon nodulaarisen amyloidoosin tyypillisiin piirteisiin.
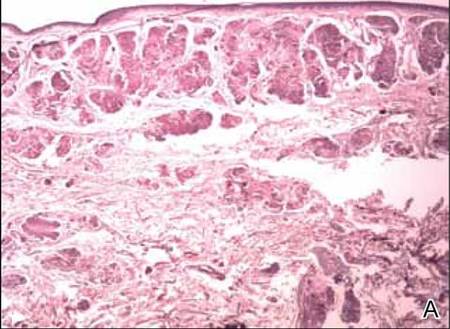
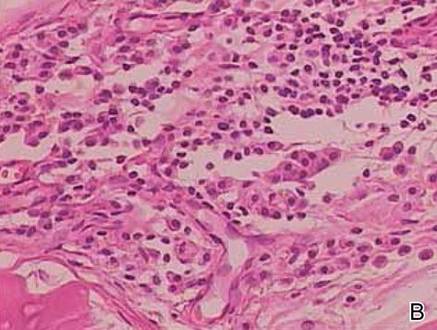
Kuva 2. Histopatologisesti todettiin retikulaariseen dermikseen ja subkutikseen kerrostunutta homogeenista eosinofiilistä materiaalia (A)(H&E, alkuperäinen suurennos ×25). Mikroskooppinen tutkimus osoitti lymfosyyttien ja plasmasolujen infiltroituneen dermikseen, erityisesti kerrostumien viereen ja verisuonten ympärille (B)(H&E, alkuperäinen suurennos ×200).
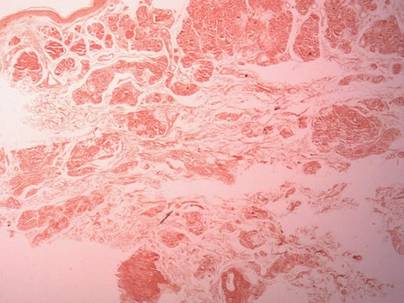
kuva 3. Kongopunavärjäys osoitti lohenpunaisen homogeenisen materiaalin (alkuperäinen suurennos ×25).
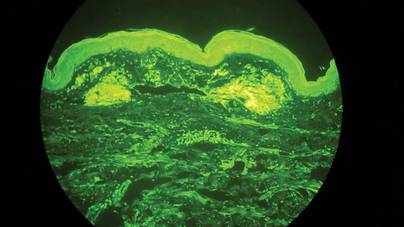
Kuva 4. Kongopunavärjäys. Tioflaviini T -värjäys osoitti kirkkaanvihreää homogeenista materiaalia (alkuperäinen suurennos ×100).
Laboratoriolöydökset
Laboratoriotutkimukset osoittivat normaalit tulokset täydellisestä verenkuvasta, virtsanäytteestä, maksan ja munuaisten toimintakokeista, veren glukoosipitoisuudesta, lipidipaneelista ja punasolujen laskeutumisnopeudesta. Seerumin proteiinielektroforeesi oli normaali, eikä Bence Jones -proteiineja havaittu. Seerumin IgA-, IgG- ja IgM-pitoisuuksissa ei havaittu poikkeavuuksia. Elektrokardiogrammi, rintakehän röntgenkuvaus ja vatsan ultraäänitutkimus olivat normaalit.
Kliinisten, histopatologisten ja laboratoriolöydösten perusteella diagnosoitiin primaarinen paikallinen ihon nodulaarinen amyloidoosi (PLCNA). Vaikka kirurgista poistoa vaiheittain ehdotettiin, potilas kieltäytyi hoidosta, koska leesiot olivat oireettomia. Ihomuutokset eivät selvästi edenneet eikä 2,5 vuoden seurannassa ollut poikkeavia systeemisiä löydöksiä.
Kommentti
Amyloidoosi on sairauksien kirjo, joka koostuu amyloidiproteiinien kertymisestä eri kudoksiin. Kliinisesti amyloidoosi jaetaan sekä primaariseen että sekundaariseen systeemiseen amyloidoosiin, hemodialyysiin liittyvään amyloidoosiin, heredofamiliaaliseen amyloidoosiin ja ihon amyloidoosiin. Primaarinen ihonalainen amyloidoosi paikallistuu iholle ilman muiden elinten osallistumista, eikä sitä esiinny systeemisessä amyloidoosissa. Systeemisen amyloidoosin sekundaarinen ihosairaus on harvinainen. Useimmat primaarisen paikallisen ihoamyloidoosin (PLCA) tapaukset ovat sporadisia, mutta noin 10 % tapauksista voi olla familiaalisia.1 Paikallisen ihoamyloidoosin kolme päämuotoa ovat makulaarinen, jäkäläinen ja nodulaarinen amyloidoosi. Nodulaarinen ihoamyloidoosi on harvinaisin PLCA:n muoto.
Nodulaarisen amyloidoosin kuvasi ensimmäisen kerran Gottron vuonna 1950.2 Sen ihomuutokset voivat esiintyä yksittäisinä tai moninkertaisina kyhmyinä, joiden päällä on toisinaan atrofisia plakkeja. Vauriot koostuvat kiinteistä, sileäpintaisista, vahamaisista tai kumimaisista, vaaleanpunaisesta ruskehtavaan vaihtelevista papuloista, plakeista tai kyhmyistä, joiden koko voi olla jopa useita senttimetrejä. Joissakin leesioissa voi näkyä pintatelangiektasiaa. On raportoitu bulloosin näköisiä ja anetodermia muistuttavia leesioita.3 Yleisin sijaintipaikka on akraalialue, jonka jälkeen tulevat jalat, pää, vartalo, käsivarret ja sukupuolielimet.4 Joissakin tapauksissa leesiot voivat parantua spontaanisti ajan myötä. Potilaallamme leesiot koostuivat sekä moninaisista kyhmyistä että atrofisista plakeista, mikä on harvinaista.
Amyloidikerrostuman patogeneesi on edelleen tuntematon. Ihon makulaarinen ja jäkäläinen amyloidoosi saattaa saada alkunsa rappeutuneista keratinosyyttien intermediaarisista filamenteista. Nodulaarinen amyloidoosi voi edustaa paikallista plasmasolujen dyskrasiaa, joka voi liittyä monoklonaaliseen gammopatiaan tai multippeliin myeloomaan.5 Joissakin PLCNA-tapauksissa amyloidin komponentit voivat koostua κ- ja λ-immunoglobuliinin kevytketjuista, ja useimmat raportoidut tapaukset ovat l-alatyyppiä.6 Erään tutkimuksen tulokset osoittivat, että β2-mikroglobuliini oli toinen amyloidifibrillien pääkomponentti ja että β2-mikroglobuliini oli osittain kehittyneen glykaation lopputuotteen modifikaation kohteena PLCNA:ssa.7
Vastaa