Hvad har DNA-minipræparater og immunudfældningseksperimenter med protein til fælles? De starter forskelligt, men de slutter med den samme, kritiske fase – eluering. Men hvad er eluering egentlig, og hvad er formålet?
Terminologien
Først skal vi starte med noget grundlæggende terminologi:
Eluering – ekstraktion af et materiale fra et andet ved at vaske det med et opløsningsmiddel.
Adsorbent – en fast fase, som kan være en silicagel i tilfælde af mini-prep-kolonner, men som regel perler, der kan være kovalent knyttet til antistoffer eller andre ligandmolekyler. “Fast fase” betyder ikke nødvendigvis en stående søjle; det kan være perler i en eppendorf, der er lette at vaske.
Affinitet – et mål for det absorberende stofs evne til at binde det valgte molekyle (det, man forsøger at eluere). Jo højere affinitet den faste fase har til det valgte biomolekyle (BOC), jo tættere binder molekylet sig til den. Man ønsker dog ikke, at bindingen skal være irreversibel; det vil gøre elueringen umulig.
Eluent – et opløsningsmiddel, der fjerner BOC fra det absorberende stof.
Eluat – det opløsningsmiddel, der indeholder BOC, som er fjernet fra det adsorberende stof.
Materialeforberedelse
For elueringen skal man absorbere det valgte molekyle, samtidig med at man fjerner forureningen. Dette er et vigtigt skridt, da den konventionelle visdom minder os om “garbage in, garbage out”. Du kan have fremragende reagenser til eluering, men hvis din prøve indeholder for meget af det uvedkommende personale (den videnskabelige betegnelse er “gunk”), vil det tilstoppe adsorptionsmaterialet. Mætning af fastfasen vil forhindre din BOC i at absorbere og derefter forurene eluatet. De effektive lyserings- og oprydningstrin er afgørende for, at dit elueringseksperiment lykkes.
Det er vigtigt at bestemme mængden af dit præ-absorptionsmateriale. Den mængde lysat, der passerer gennem absorptionsmediet, bør ikke overstige 3 – 5 volumener af kolonnen. Den store mængde lysat, der passerer gennem absorptionsmidlet, forlænger forsøgets varighed og øger sandsynligheden for absorption af gunk. I mange tilfælde kan det betale sig at reducere det oprindelige lysatvolumen ved filtrering eller fraktionering. Lysatvolumen bestemmer således kolonnens størrelse.
Valget af adsorptionsmateriale afhænger af den kemiske sammensætning af dit interessemolekyle. Absorptionen af biomolekyler indebærer normalt mere eller mindre specifik interaktion mellem substratet og molekylet. F.eks. absorberes DNA på minikolonner på grund af ionisk interaktion mellem negativt ladede DNA-fosfatgrupper og positivt ladede silica-partikler.
Proteiner adsorberes normalt på sepharose eller magnetiske perler dækket af IgG.
Efter en indledende lysatpåføring kan din kolonne på intet tidspunkt tørre ud. Dette vil “bage dit” molekyle til det absorberende stof og forstyrre kolonneintegriteten. Hvis du ikke har tid til at fortsætte forsøget, skal du fylde kolonnen op med en kompatibel buffer og stoppe flowet.
Vaskning
Sigtet med vask af den faste fase er at fjerne et ikke-relateret materiale, mens det interessante molekyle forbliver på kolonnen. Den selektive adskillelse opnås ofte ved at anvende en buffer med lav ionisk styrke (f.eks. lav saltkoncentration). Volumenet af vaskebufferen bør ligge tæt på mængden af det oprindelige materiale og være mindst 3-5 volumener af kolonnen.
Når flere volumener af vaskebufferen er passeret gennem kolonnen, vil forureningerne imidlertid være vasket væk, og yderligere vask vil ikke forbedre kvaliteten af din prep. Desuden vil du begynde at miste dit målmateriale.
Elution
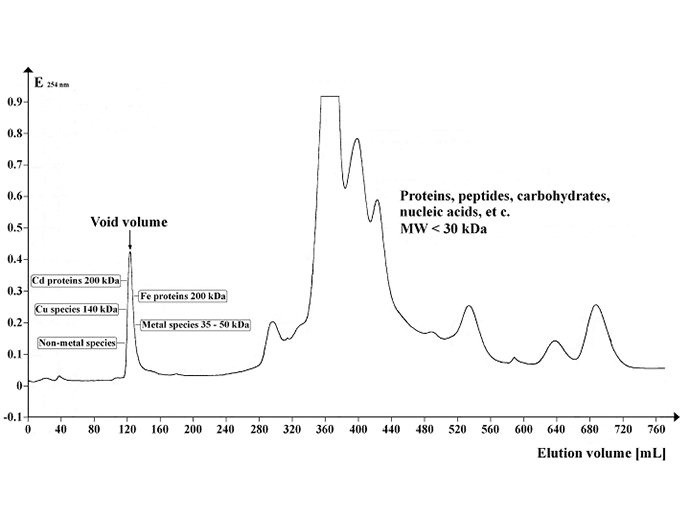
Billede: Kromatogram, der viser UV-absorptionsprofilen af Arabidopsis-supernatant, der er separeret på Sephadex G-50 Superfine. Gelvolumen: 500 mL; søjlelængde: 700 mm; søjlediameter: 30 mm; elueringsmiddelstrømningshastighed: 12 mL / time; fraktionsvolumen: 8,0 mL; antal fraktioner: 95; prøvevolumen: 5 mL; separationstemperatur: 4 °C; elueringsbuffer: 20 mM Tris-HCl, 1 mM NaN3; pH 8,0. Billedkredit: https://commons.wikimedia.org/wiki/File:Chrom_SephG-50.tif
Elution i sig selv fungerer, fordi du afbryder bindingerne mellem søjlen og substratet (dvs. ved at bruge højt salt eller høj temperatur på eluenten). Elution sker normalt i et lille volumen buffer, der er foreneligt med opbevaring af prøven og yderligere anvendelser.
DNA-elution fra mini-prep-kolonnen er det enkleste tilfælde: et volumen buffer fjerner næsten alt DNA. Koncentrationen af DNA i eluatet er omvendt proportional med den anvendte elueringsbuffer: jo mere buffer man bruger, jo mindre er den endelige DNA-koncentration. Selv i dette tilfælde anbefaler de fleste producenter dog, at der anvendes et ekstra volumen for at fjerne alt DNA.
For kolonnerne er elueringshastigheden kritisk. For langsom hastighed vil øge risikoen for nedbrydning af molekylet; for hurtig, og der vil ikke være nogen opløsning af fraktioner.
For kolonner med stort volumen skal du indsamle eluatfraktioner, fordi dit molekyle vil være fordelt mellem dem. Den første fraktion vil indeholde en blanding af vaske- og elueringsbuffer og eventuel kontaminering, der ikke fjernes af vaskebufferen.
Du kan overvåge OD for din type molekyle (260nm/280nm for DNA) og lave en blot for din specifikke molekylkoncentration i hver fraktion. I det enkleste tilfælde vil din molekylefordeling følge en simpel klokkekurve, men den kan have en eller flere skarpe toppe.
Sluttelig vil du, hvis du kender de grundlæggende parametre for dit forsøg (absorbent, kolonnestørrelse, vaskebuffer, elueringsbuffer, flowhastighed, antal fraktioner) og de generelle principper for eluering, kunne opstille din eluering med succes.
For at få flere detaljer skal du finde en artikel, hvor de andre forskere har gjort noget lignende – ideelt set det samme molekyle, men et lignende vil være tilstrækkeligt – og tilpasse det til dine forhold.
Skriv et svar